AI솔루션기업, AI소프트웨어, AI프로그램개발, AI서비스개발을 통해 AI개발업체로서 혁신을 선도합니다.
-
AIDESIGN
데이터 주도 AI솔루션기업으로서 AI기업, AI소프트웨어, AI솔루션개발,
AI프로그램개발을 통해 복잡한 비즈니스 난제를 해결하고 AI서비스개발을 선도합니다.As a data-driven AI solutions company, we lead AI enterprise initiatives—providing AI software,
AI solution development, and AI program development—to solve complex business challenges
and pioneer AI service development. -

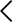 Back
Back생성형AI 의료기기, 韓 식약처가 FDA보다 먼저 승인한 이유는?
생성형AI 의료기기, 韓 식약처가 FDA보다 먼저 승인한 이유는? - > "규제가 혁신을 막는다"는 공식이 깨졌다. 이번엔 규제가 혁신을 이끌었다.

美 FDA도 못 한 일을 韓 식약처가 해냈다 — 생성형 AI 의료기기, 세계 최초 승인의 진짜 의미
> "규제가 혁신을 막는다"는 공식이 깨졌다. 이번엔 규제가 혁신을 이끌었다.
---
---
핵심 요약
2026년 현재, 한국 식품의약품안전처(식약처)가 전 세계 최초로 생성형 AI 기반 디지털 의료기기를 정식 승인했다.
흉부 X-ray 영상을 분석해 57종 이상 소견을 텍스트 예비소견서로 자동 작성하는 소프트웨어가 3등급 의료기기로 허가를 받은 것이다.
미국 FDA는 현재 1,450여 개의 AI·ML 의료기기를 승인했음에도, 생성형 AI를 임상 의사 결정에 직접 활용하는 사례는 단 한 건도 없다.
한국이 이 보수적 장벽을 먼저 넘을 수 있었던 핵심은 세계 최초 '생성형 AI 의료기기 허가·심사 가이드라인'의 선제적 발간이다.
이번 승인은 단순한 국내 이슈를 넘어, 글로벌 의료 AI 표준을 누가 주도하느냐는 게임의 판도를 바꾸는 사건이다.
---
심층 분석 — 왜 이 뉴스가 중요한가
---
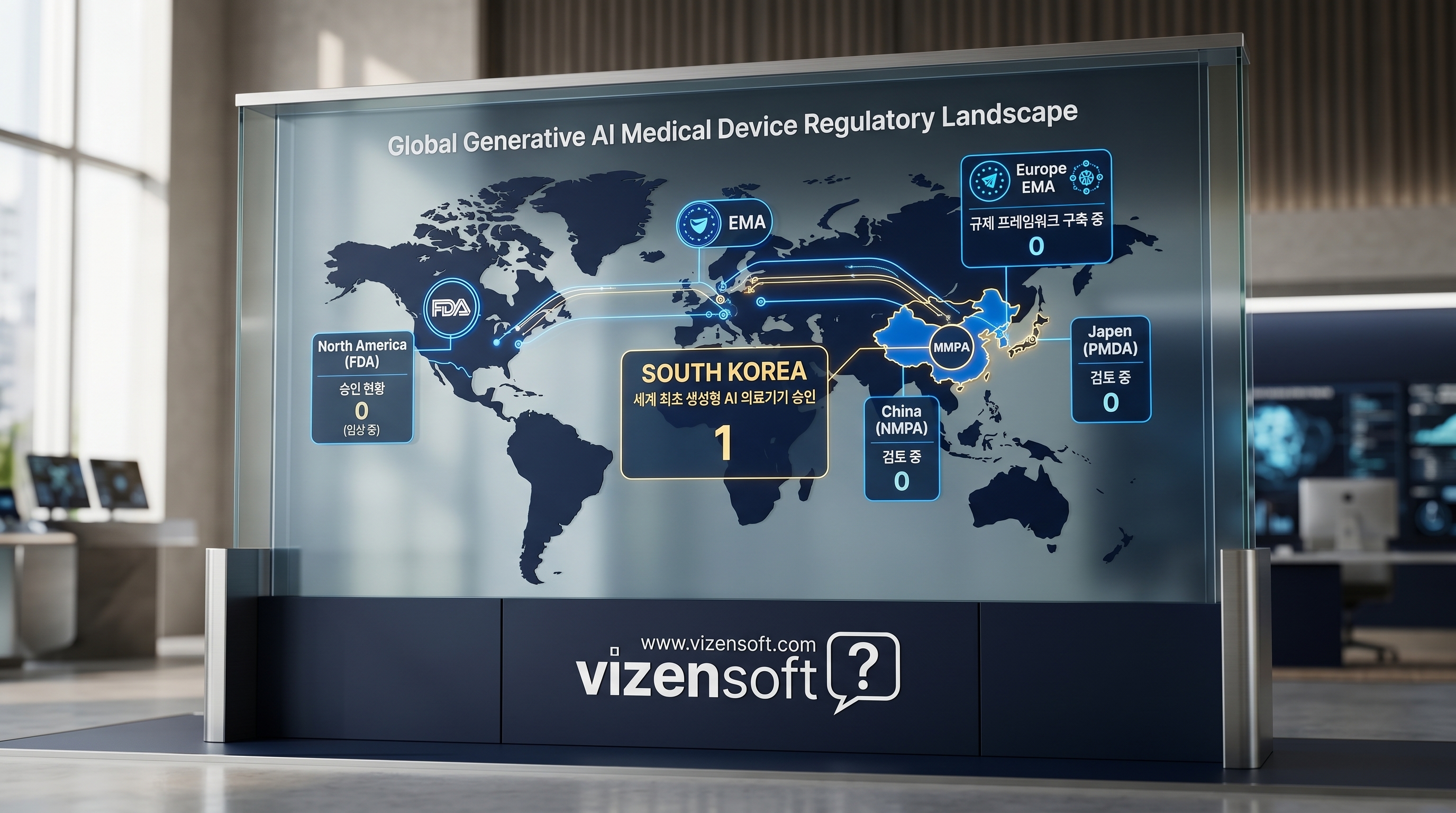
의료 AI의 역사는 크게 두 시대로 나뉜다.
첫 번째 시대는 고정된 데이터셋 안에서 이미지를 분류하거나 이상 부위를 표시하는 예측·판독형 AI의 시대다.
FDA가 승인한 1,450여 개의 AI 의료기기 대부분이 이 범주에 속한다.
입력과 출력이 고정적이므로 기존 심사 틀, 즉 동등성 입증 방식으로 평가가 가능했다.
두 번째 시대는 지금 시작되고 있다.
대형언어모델(LLM)을 기반으로 자유로운 텍스트를 생성하는 생성형 AI가 임상 현장에 진입하는 시대다.
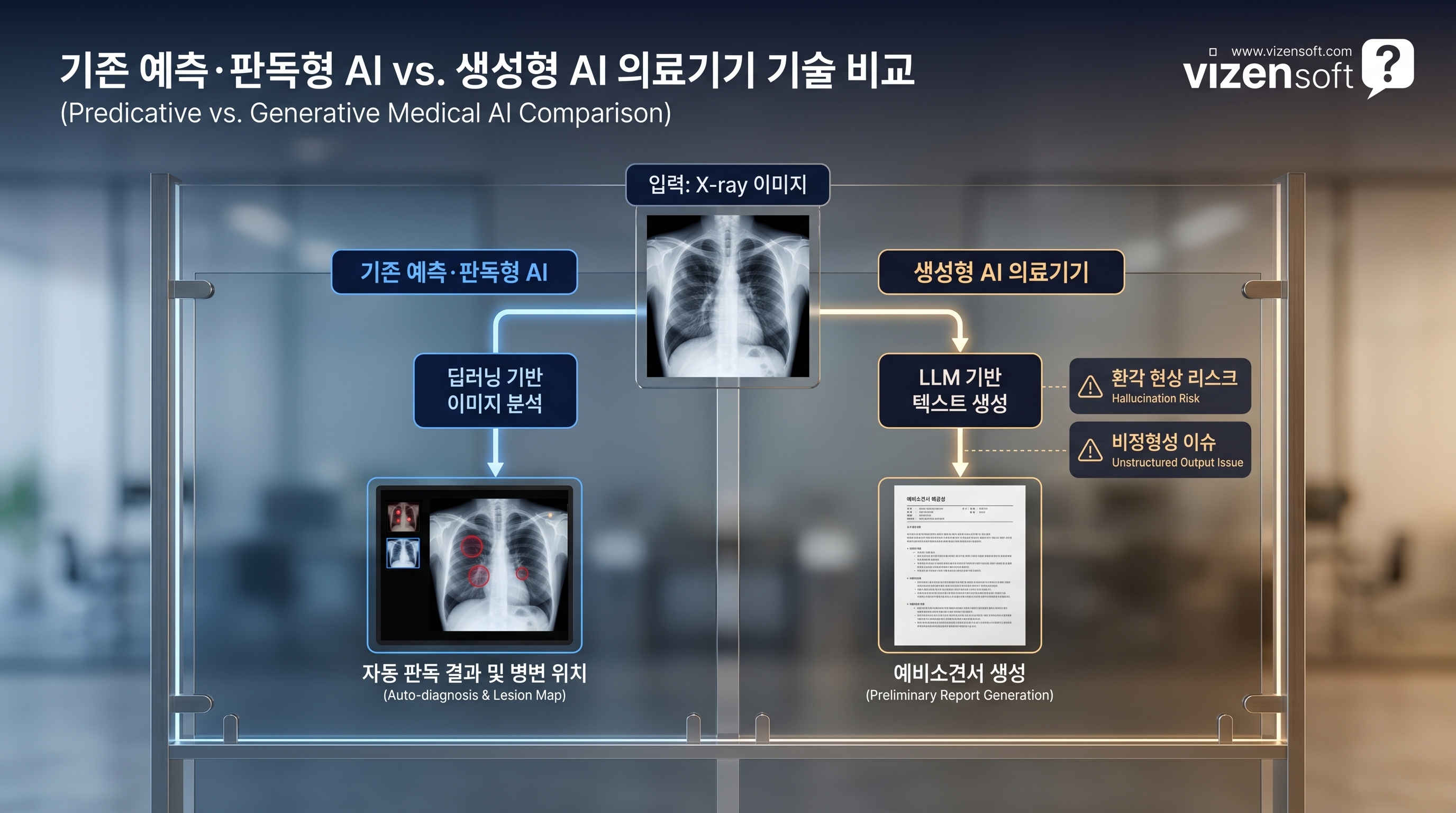
이 단계에서 규제 당국이 직면하는 두 가지 근본적 난제가 있다.
① 환각 현상(Hallucination) — AI가 존재하지 않는 소견을 그럴듯하게 생성할 수 있다는 위험
② 비정형성 — 동일한 입력에도 매번 결과값이 달라질 수 있어 재현성 검증이 어렵다는 문제
FDA가 전담 위원회를 신설하고도 아직 명확한 심사 트랙을 완성하지 못한 이유가 바로 여기에 있다.
기존의 심사 프레임워크로는 생성형 AI의 안전성을 평가하는 것 자체가 불가능에 가까웠던 것이다.
---
---
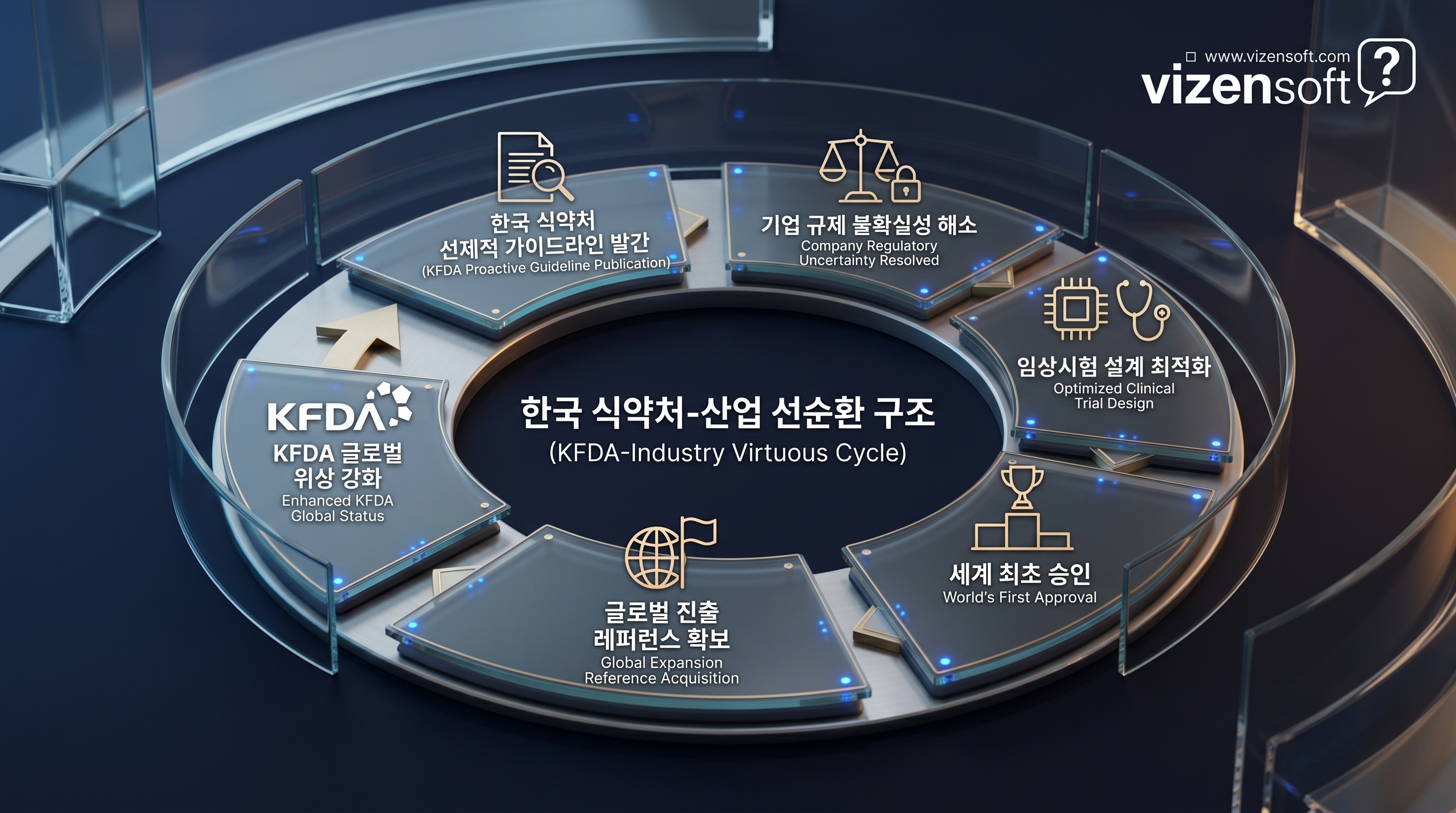
회사의 견해 — 규제가 '장벽'에서 '무기'가 된 순간
---
이번 사건의 핵심을 한 문장으로 정리하면 이렇다.
"한국은 혁신을 허용한 것이 아니라, 혁신이 가능한 판을 먼저 만들었다."
식약처가 '디지털의료제품법' 시행에 발맞춰 세계 최초로 생성형 AI 전용 허가·심사 가이드라인을 발간한 것은 단순한 행정 절차가 아니다.
이는 규제 당국이 산업보다 한발 앞서 미래를 정의한 행위다.
우리가 주목하는 것은 이 구조적 전환이 가져오는 세 가지 함의다.
첫째, '퍼스트 무버 어드밴티지'의 재정의다.
기술에서의 퍼스트 무버가 아닌 규제에서의 퍼스트 무버가 글로벌 스탠더드를 결정하는 시대가 열렸다.
한국이 설계한 임상 평가 기준과 허가 프레임이 향후 FDA, EMA의 참조 모델이 될 가능성이 높다.
둘째, 국내 승인이 곧 글로벌 레퍼런스가 된다.
까다롭기로 정평이 난 한국 식약처의 3등급 승인을 획득했다는 사실은, 기업이 해외 규제 기관과 협의할 때 강력한 신뢰 자산으로 작동한다.
글로벌 진출 비용과 시간을 획기적으로 단축할 수 있는 레버리지다.
셋째, AI 헬스케어 생태계 전반의 기준점이 설정된다.
생성형 AI 의료기기의 임상시험 설계, 안전성 평가, 사후 모니터링 체계가 공식화됨으로써, 후발 기업들도 명확한 로드맵 위에서 개발을 진행할 수 있게 된다.
---

---
기업·개발자에게 미치는 영향

---
이번 승인이 AI 헬스케어 관련 기업과 개발자에게 미치는 실질적 영향은 다음과 같이 정리된다.
1. 임상시험 설계의 패러다임 전환
생성형 AI의 비정형 출력을 평가하는 새로운 임상 프로토콜이 공식화됐다.
앞으로 LLM 기반 의료 소프트웨어를 개발하는 모든 기업은 이 프레임을 기준으로 임상을 설계해야 한다.
2. 규제 전략의 우선순위 재편
"기술을 먼저 개발하고 규제를 나중에 맞추는" 방식은 더 이상 통하지 않는다.
규제 가이드라인을 개발 초기 단계부터 내재화하는 Regulatory-by-Design 접근이 필수가 된다.
3. 글로벌 진출 시퀀스의 변화
과거에는 FDA 승인 후 한국 진출이 일반적이었다.
이제는 한국 식약처 승인 → 글로벌 레퍼런스 확보 → FDA·EMA 협상이라는 새로운 시퀀스가 현실적 옵션이 된다.
4. 사후 안전성 모니터링 체계의 중요성 급부상
승인 이후의 실사용 데이터 수집과 안전성 모니터링이 허가 유지 및 글로벌 신뢰 확보의 핵심 변수로 부상한다.
---

---
향후 전망 및 제언

---
이번 한국의 세계 최초 승인은 시작점이지 완성점이 아니다.
향후 전망과 업계가 준비해야 할 사항을 짚는다.
먼저, 글로벌 규제 수렴이 가속화될 것이다.
FDA와 EMA는 한국의 사례를 면밀히 분석할 것이고, 이를 참조한 자체 가이드라인을 2~3년 내 완성할 가능성이 높다.
이 기간이 국내 기업이 글로벌 시장에서 선점 우위를 확보할 수 있는 골든 윈도우다.
다음으로, 사후 모니터링이 다음 전쟁터가 된다.
생성형 AI 의료기기의 신뢰는 승인 이후 실사용 환경에서 쌓인다.
환각 발생률, 전문의 검토 필요 케이스 비율, 임상 결과 개선 지표 등의 리얼월드 데이터(RWD) 축적이 다음 단계 경쟁력을 결정한다.
그리고, AI 기술 기업과 의료 기관의 협력 모델이 진화해야 한다.
기술을 만드는 기업과 이를 검증하는 의료 기관 사이의 데이터 공유, 공동 임상 설계, 피드백 루프 구축이 생태계 경쟁력의 핵심이 될 것이다.
마지막으로, 우리의 제언이다.
AI 헬스케어를 준비하는 기업이라면, 지금 당장 식약처의 생성형 AI 의료기기 허가·심사 가이드라인을 정독할 것을 권고한다.
이 문서는 단순한 규제 지침서가 아니라, 글로벌 AI 의료 시장의 첫 번째 공식 룰북이다.
---

---

---
> vizensoft AI 분석 인사이트
> 기술의 경쟁력은 코드에만 있지 않다.
> 규제를 설계하는 능력, 그 규제 안에서 가장 먼저 움직이는 실행력이 AI 시대의 진짜 해자(moat) 다.
> 한국이 이번에 증명한 것이 바로 그것이다.
---
🏢 **vizensoft** | AI 기술 전문 분석
📧 sales@vizensoft.com | 📞 02-338-4610
AI가 의료 현장의 언어를 말하기 시작했습니다. 그 첫 문장을 한국이 썼습니다. 다음 챕터를 함께 준비하세요 🚀
🔗 https://www.vizensoft.com
---
**#생성형AI의료기기 #식약처세계최초승인 #AI헬스케어 #디지털의료기기 #AIMD #의료AI규제 #LLM의료 #FDA비교 #디지털헬스케어 #AIRead**






